Specialty Drug Development Process
By understanding how biologics are developed, actuaries can help manage this growing area of cost
February 2021Photo: Shutterstock/D. Kucharski K. Kucharska
Specialty pharmacy is among the fastest-growing segments of health care costs. It increased by $54 billion from 2011 to 2016 and accounted for 73 percent of all medicine spending growth during that time. In 2020, specialty pharmacy spend was nearly 50 percent of all prescription drug spend.1
The process used to develop traditional drugs was discussed in “Traditional Drug Development Process” in May 2020. As a follow-up, this article provides background on the research and development of specialty drugs in contrast to traditional drugs. Unfortunately, there is no single definition for specialty drugs (while biologic drugs typically are considered specialty drugs, specialty drugs are not always biologics).2 For the purpose of this article, we will focus primarily on the research and discovery of biologics.
Biological products are large, complex molecules—particularly when compared to the chemicals used to make traditional drugs—and biologics often are referred to as “large molecules.” A living system such as an animal or animal cell is used in the production of a biologic. Biologics include a wide range of products such as vaccines, blood and blood components, allergenics, somatic cells, gene therapy, tissues and recombinant therapeutic proteins.3 As we review the development process of biologics, we will break it down based on common subcategories of biologic drugs, including monoclonal antibodies, gene therapy and cell therapies (see Figure 1).
Figure 1: Specialty Drugs and Biologics
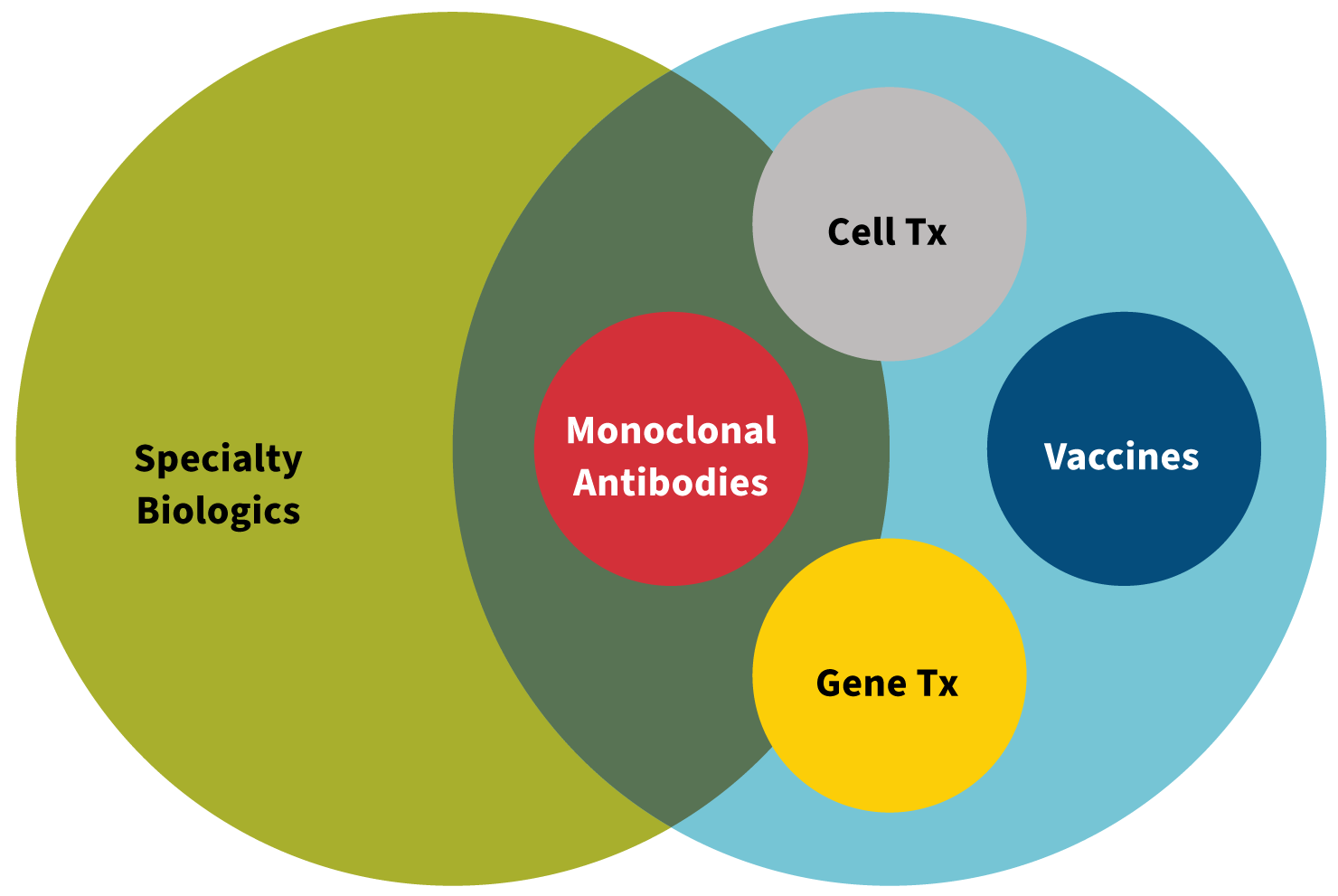
Source: Created by Kerri Miller, Pharm.D.
In contrast to traditional drug development, biologic drug development requires the use of aseptic handling techniques starting with the initial manufacturing steps. Following aseptic techniques refers to the sterile environment and processing used in the manufacturing of biologics. This process minimizes contamination to the final product, which could result in infection and/or poor outcomes for the end user.
Biologics also differ from traditional drugs in that biologics are not manufactured into pills for patients to swallow, but rather they are administered to a patient traditionally via an injection under the skin (aka subcutaneous or SubQ) or via an infusion directly into a vein (aka intravenous or IV). The route of administration adds a new dimension to the skill required by either the patient or a caregiver as well as different safety and side effects that can come with these types of administration. This leads to new and unique considerations in the manufacturing process of biologics compared to traditional drugs.
Monoclonal Antibodies
The concept and utility of monoclonal antibody development exploded in the 1970s. Georges Kohler and Cesar Milstein successfully fused myeloma cell lines with B cells and created hybridomas that ultimately led to monoclonal antibodies (see Figure 2). The hybridoma process starts by injecting a mouse with a specific protein or antigen, which then stimulates the immune response in the mouse. Antigen-specific antibody-producing spleen B cells from the mouse are fused with myeloma cells, which give the new hybridoma the longevity from the myeloma cell and the antibody-producing capability from the spleen cell. The hybridomas can be grown in cultures in the laboratory, and the monoclonal antibody can be purified from the culture medium.
Figure 2: Producing Monoclonal Antibodies
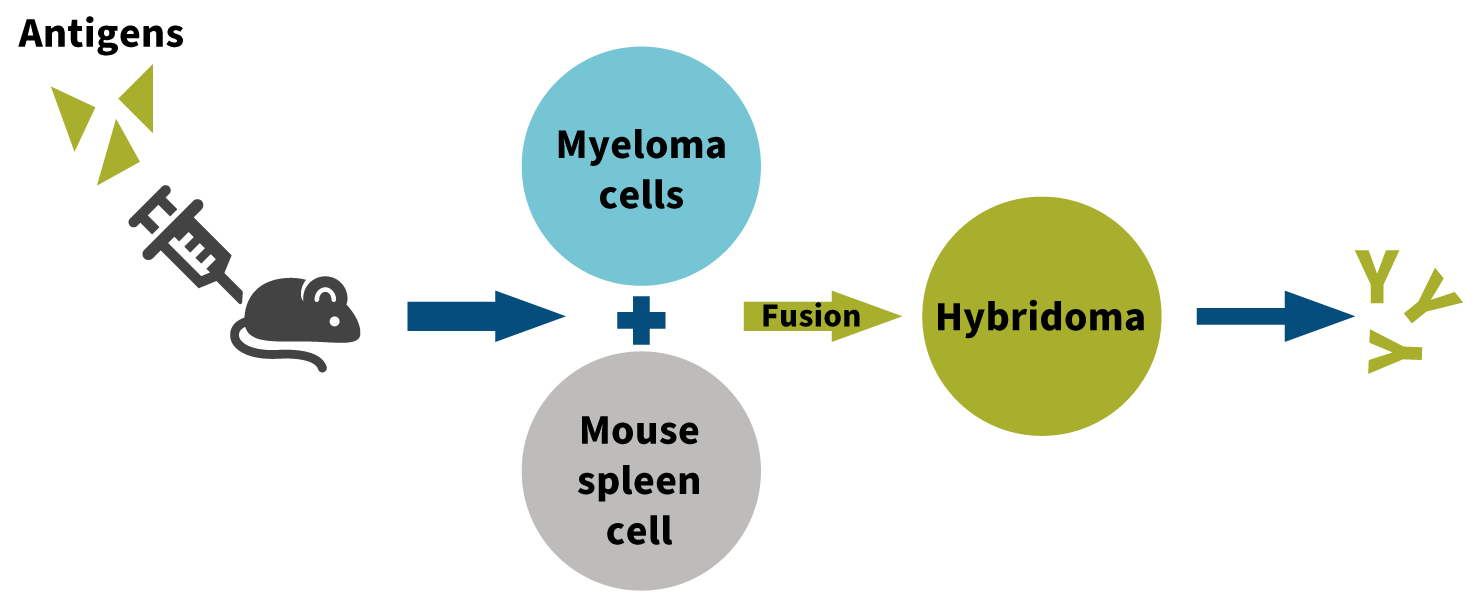
Source: Created by Kerri Miller, Pharm.D.
Besides the hybridoma technique, other approaches to isolate monoclonal antibodies have been developed.4 For example, newer technology called antibody-drug conjugates (ADCs) combine the targeting capabilities of monoclonal antibodies with the cancer-killing ability of cytotoxic drugs.5 Of the more than 80 monoclonal antibodies approved by the U.S. Food and Drug Administration (FDA), more than 90 percent are generated by the traditional hybridoma technology.6 Some of the first monoclonal antibodies the FDA approved in the late 1990s include Rituxan, Remicade and Herceptin.
Biologics License Application Filing with the FDA
Whereas traditional drugs require a new drug application (NDA) for approval, biologics require a biologics license application (BLA) to be filed with the FDA. In addition to using the same FDA-provided form, NDAs and BLAs share common features:
- They have a fast-track designation.
- They require financial disclosure.
- There are labeling and advertising regulations.
- There are pediatric study requirements.
- They allow for accelerated approval.
- There is orphan drug exclusivity.7,8
Despite these common features, significant differences exist. Only NDAs provide for patent and pediatric exclusivity. NDAs have more extensive regulations that pertain to content and filing criteria.9 On the other hand, BLA regulations largely focus on the manufacturing process and facility. For BLA approval, the manufacturing process and facility must satisfy safety, purity and potency requirements.10
505(b)(2) Regulatory Pathway
Like biologic drugs, biosimilars are composed of complex molecules manufactured from living organisms. The FDA defines a biosimilar as “a biological product that is highly similar to and has no clinically meaningful differences” compared to an approved biologic drug.11 The biologic drug to which the biosimilar is compared is known as the “reference product.” The manufacturer of the biosimilar must demonstrate that the structure and function of the biosimilar is highly similar to the reference product and that there are no clinically meaningful differences in terms of safety, purity and potency.12
Biosimilars are not necessarily interchangeable with the corresponding reference products, however. A biosimilar drug is interchangeable with the reference product if and only if the biosimilar meets the two additional requirements stipulated by the Biologics Price Competition and Innovation Act:
- The biosimilar has been demonstrated to produce the same clinical result as the reference product.
- The risk of alternating or switching between using the biosimilar and the reference product is not greater than the risk of only using the reference product.13
If the FDA determines the biosimilar is interchangeable with the reference product, the biosimilar may be substituted for the biologic drug without the prescriber’s approval.14 Otherwise, the prescriber’s approval is required before a biosimilar can be substituted for a biologic.
On the surface, it may appear that a biosimilar is simply a generic version of a biologic drug. However, it’s important to note that a biosimilar is not equivalent to a generic drug. Biosimilars and generics are approved by the FDA through different abbreviated pathways that do not require the manufacturer to duplicate expensive clinical trials.15 (Biosimilars are approved through the 505(b)(2) pathway, whereas generics are approved using an abbreviated new drug application.)
The difference between a biosimilar and a generic is in the details of what is required for FDA approval. For a generic drug, the active ingredients must be identical to the brand-name drug, and the generic manufacturer must demonstrate bioequivalency.
Real-Time Oncology Review
The FDA currently is piloting a real-time oncology review (RTOR) program. The goal of the RTOR pilot program is to create a more efficient and timely review process for “drugs likely to demonstrate substantial improvements over available therapy.”16
The RTOR program is designed to allow the FDA to begin reviewing data before the manufacturer actually submits the NDA or BLA. By reviewing data prior to the submission of the complete application, the FDA can provide feedback to the manufacturer regarding data quality issues, data analysis methods and review questions. The manufacturer is then able to incorporate this feedback into the application, which allows for a more efficient and timely review by the FDA.
Specialty Drug Cost
The average monthly supply for a traditional brand drug is several hundred dollars, while the cost is several thousand dollars for a specialty brand drug. Even though only 2 percent of the population utilizes specialty drugs, they account for nearly half of the drug spend.17 Specialty drug share of net spending across institutional and retail settings rose from 26.2 percent in 2009 to 49.5 percent in 2018.18 As an example, Mavenclad, a drug that treats multiple sclerosis, has a wholesale acquisition cost (WAC) of $99,500 annually (note that WAC is usually not the actual price paid).19
Being designated as a specialty drug can be useful to help control costs. Health plans often have a different tier in the formulary for specialty drugs, which determines member cost-share and may incentivize members to seek more cost-effective treatments if possible. Health plans and pharmacy benefits managers (PBMs) often will negotiate drug prices with pharmacies on an aggregate level for nonspecialty drugs. But given the higher price and complexity of specialty drugs, these prices may be negotiated on an individual drug level.
Actuaries Can Play a Role
Actuaries already contribute to specialty drug cost trend management through traditional drug benefit plan design strategies and by evaluating the potential cost impact of different specialty pharmacy distribution and utilization and care management programs. Continued innovation in this area and more specific therapy-level actuarial involvement can further help ensure patients receive the innovative care they need at an affordable cost-share and premium and employee contribution rate levels.
Statements of fact and opinions expressed herein are those of the individual authors and are not necessarily those of the Society of Actuaries or the respective authors’ employers.
References:
- 1. Advanced Medical Reviews. The Rising Cost of Speciality Drugs. Prescription Drugs, January 15, 2020 (accessed June 2, 2020). ↩
- 2. Kirchhoff, Suzanne M. Speciality Drugs: Backgrounds and Policy Concerns. Congressional Research Service, 26. August 3, 2015 (accessed May 15, 2020). ↩
- 3. U.S. Food & Drug Administration. Developing Products for Rare Diseases & Conditions. U.S. Food & Drug Administration, December 20, 2018 (accessed December 11, 2020). ↩
- 4. Ho, Mitchell. 2018. Inaugural Editorial: Searching for Magic Bullets. Antibody Therapeutics, 1, no. 1:1-5. ↩
- 5. Wikipedia contributors. Antibody-drug Conjugate. Wikipedia, September 27, 2020 (accessed October 3, 2020). ↩
- 6. Zaroff, S., and G. Tan. 2019. Hybridoma Technology: The Preferred Method for Monoclonal Antibody Generation for In Vivo Applications. Biotechniques 67, no. 3:90-92. ↩
- 7. U.S. Food & Drug Administration. Therapeutic Biologics Applications (BLA). U.S. Food & Drug Administration, February 24, 2020 (accessed April 8, 2020). ↩
- 8. U.S. Food & Drug Administration. New Drug Applications (NDA) vs. Biologics License Applications (BLA). U.S. Food & Drug Administration (accessed April 8, 2020). ↩
- 9. Ibid. ↩
- 10. Supra note 8. ↩
- 11. U.S. Food & Drug Administration. Biosimilar and Interchangeable Products. U.S. Food & Drug Administration, October 23, 2017 (accessed March 17, 2020). ↩
- 12. Ibid. ↩
- 13. U.S. Food & Drug Administration. Implementation of the Biologics Price Competition and Innovation Act of 2009. U.S. Food & Drug Administration, February 12, 2016 (accessed March 17, 2020). ↩
- 14. Supra note 11. ↩
- 15. Ibid. ↩
- 16. U.S. Food & Drug Administration. Real-Time Oncology Review Pilot Program. U.S. Food & Drug Administration, October 7, 2020 (accessed July 10, 2020). ↩
- 17. Express Scripts. 2019 Drug Trend Report. Evernorth Health Inc. (accessed December 11, 2020). ↩
- 18. The IQVIA Institute. Medicine Use and Spending in the U.S. Institute Report, May 9, 2019 (accessed December 11, 2020). ↩
- 19. National Multiple Sclerosis Society. FDA Approves Cladribine–Brand Named Mavenclad—for People With Relapsing-Remitting MS and Active Secondary Progressive MS—Update. National Multiple Sclerosis Society, April 2, 2019 (accessed December 11, 2020). ↩
Copyright © 2021 by the Society of Actuaries, Chicago, Illinois.